All-electron real-time and imaginary-time time-dependent density functional theory within a numeric atom-centered basis function framework
Abstract
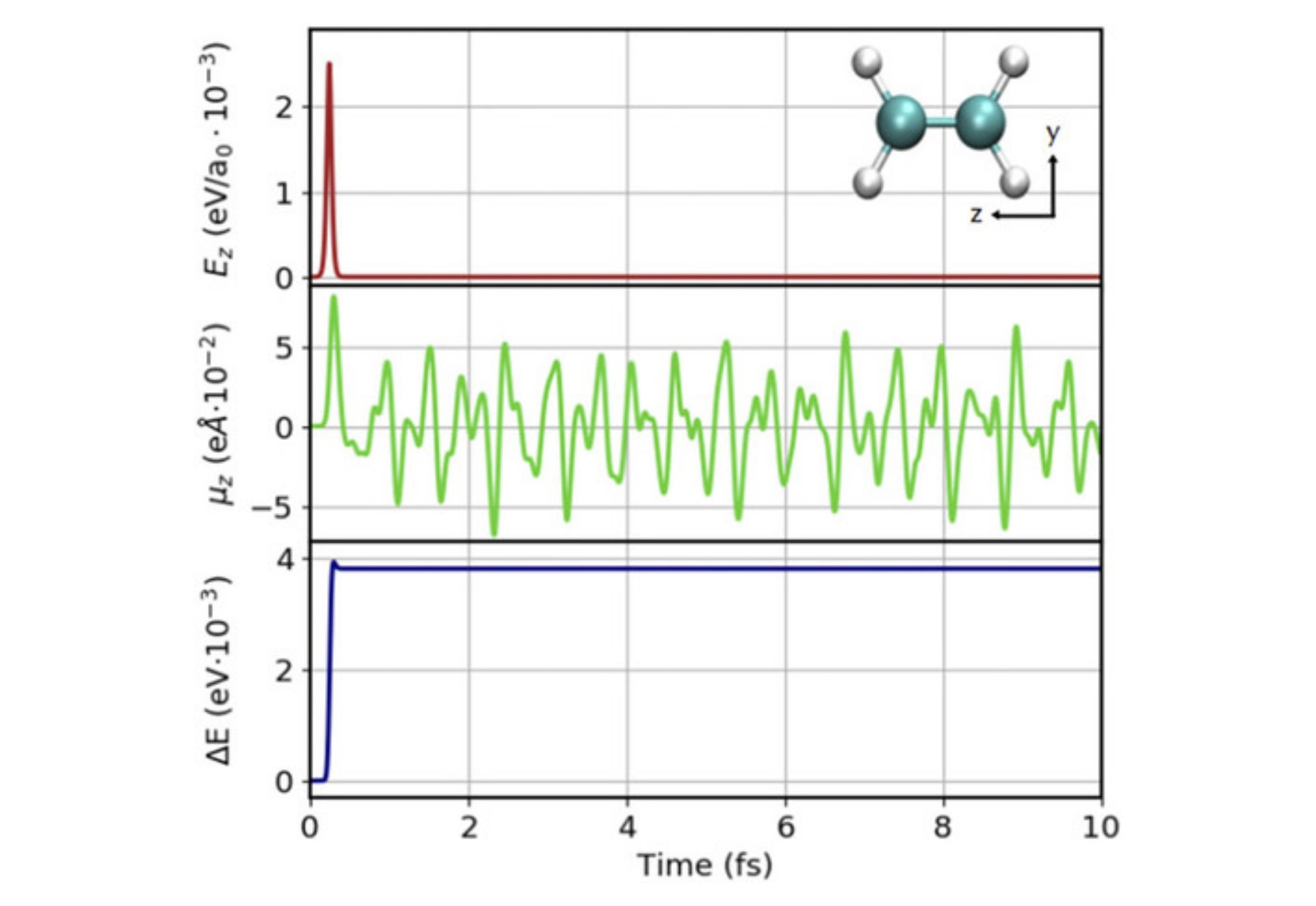
Real-time time-dependent density functional theory (RT-TDDFT) is an attractive tool to model quantum dynamics by real-time propagation without the linear response approximation. Sharing the same technical framework of RT-TDDFT, imaginary-time time-dependent density functional theory (it-TDDFT) is a recently developed robust-convergence ground state method. Presented here are high-precision all-electron RT-TDDFT and it-TDDFT implementations within a numerical atom-centered orbital (NAO) basis function framework in the FHI-aims code. We discuss the theoretical background and technical choices in our implementation. First, RT-TDDFT results are validated against linear-response TDDFT results. Specifically, we analyze the NAO basis sets’ convergence for Thiel’s test set of small molecules and confirm the importance of the augmentation basis functions for adequate convergence. Adopting a velocity-gauge formalism, we next demonstrate applications for systems with periodic boundary conditions. Taking advantage of the all-electron full-potential implementation, we present applications for core level spectra. For it-TDDFT, we confirm that within the all-electron NAO formalism, it-TDDFT can successfully converge systems that are difficult to converge in the standard self-consistent field method. We finally benchmark our implementation for systems up to ∼500 atoms. The implementation exhibits almost linear weak and strong scaling behavior.
Citation
Joscha Hekele, Yi Yao, Yosuke Kanai, Volker Blum, and Peter Kratzer, "All-electron real-time and imaginary-time time-dependent density functional theory within a numeric atom-centered basis function framework", J. Chem. Phys. 155, 154801 (2021) https://doi.org/10.1063/5.0066753