All-Electron BSE@GW Method for K-Edge Core Electron Excitation Energies
Abstract
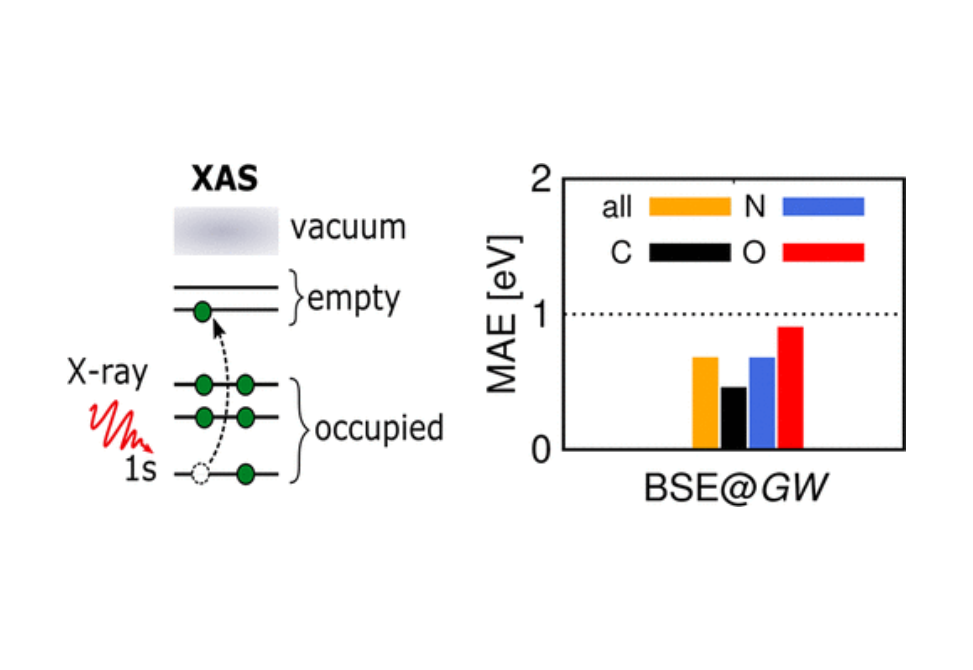
We present an accurate computational approach to calculate absolute K-edge core electron excitation energies as measured by X-ray absorption spectroscopy. Our approach employs an all-electron Bethe–Salpeter equation (BSE) formalism based on GW quasiparticle energies (BSE@GW) using numeric atom-centered orbitals (NAOs). The BSE@GW method has become an increasingly popular method for the computation of neutral valence excitation energies of molecules. However, it was so far not applied to molecular K-edge excitation energies. We discuss the influence of different numerical approximations on the BSE@GW calculation and employ in our final setup (i) exact numeric algorithms for the frequency integration of the GW self-energy, (ii) G0W0 and BSE starting points with ∼50% of exact exchange, (iii) the Tamm–Dancoff approximation and (iv) relativistic corrections. We study the basis set dependence and convergence with common Gaussian-type orbital and NAO basis sets. We identify the importance of additional spatially confined basis functions as well as of diffuse augmenting basis functions. The accuracy of our BSE@GW method is assessed for a benchmark set of small organic molecules, previously used for benchmarking the equation-of-motion coupled cluster method [Peng et al., J. Chem. Theory Comput., 2015, 11, 4146], as well as the medium-sized dibenzothiophene (DBT) molecule. Our BSE@GW results for absolute excitation energies are in excellent agreement with the experiment, with a mean average error of only 0.63 eV for the benchmark set and with errors <1 eV for the DBT molecule.
Citation
All-Electron BSE@GW Method for K-Edge Core Electron Excitation Energies
DOI: 10.1021/acs.jctc.1c01180